Alcoholic ketoacidosis (AKA) is a metabolic disorder commonly observed in chronic alcohol users, characterized by the accumulation of ketone bodies due to impaired glucose utilization and increased fatty acid metabolism. One of the key metabolic cofactors involved in these processes is nicotinamide adenine dinucleotide (NADH), which plays a critical role in redox reactions and energy production. In the context of AKA, the relationship between NADH levels and the pathophysiology of the condition is of particular interest. Elevated NADH levels are often associated with increased ethanol metabolism, as alcohol dehydrogenase converts ethanol to acetaldehyde, generating NADH in the process. This excess NADH can disrupt the NAD+/NADH ratio, impairing key metabolic pathways such as the citric acid cycle and contributing to the development of ketoacidosis. Thus, understanding whether NADH is elevated in alcoholic ketoacidosis is essential for elucidating the metabolic derangements underlying this condition and potentially identifying therapeutic targets.
| Characteristics | Values |
|---|---|
| NADH Levels in Alcoholic Ketoacidosis | Elevated |
| Mechanism of Elevation | Increased ethanol metabolism depletes NAD+ and increases NADH levels |
| Role of Ethanol Metabolism | Ethanol is converted to acetaldehyde by alcohol dehydrogenase, consuming NAD+ and producing NADH |
| Clinical Significance | Elevated NADH contributes to metabolic acidosis and impaired gluconeogenesis |
| Impact on Lactic Acid Production | Elevated NADH promotes reduction of pyruvate to lactate, increasing lactate levels |
| Diagnostic Relevance | Elevated NADH is a marker of alcohol-induced metabolic disturbances |
| Therapeutic Implications | NAD+ replenishment may be considered in severe cases to restore redox balance |
| Comparison to Diabetic Ketoacidosis | In DKA, NADH levels are typically not elevated |
| Laboratory Findings | Elevated serum lactate, ketones, and anion gap with normal glucose |
| Patient Population | Chronic alcohol users with poor nutritional status |
Explore related products






What You'll Learn
![]()
NADH Role in Metabolism
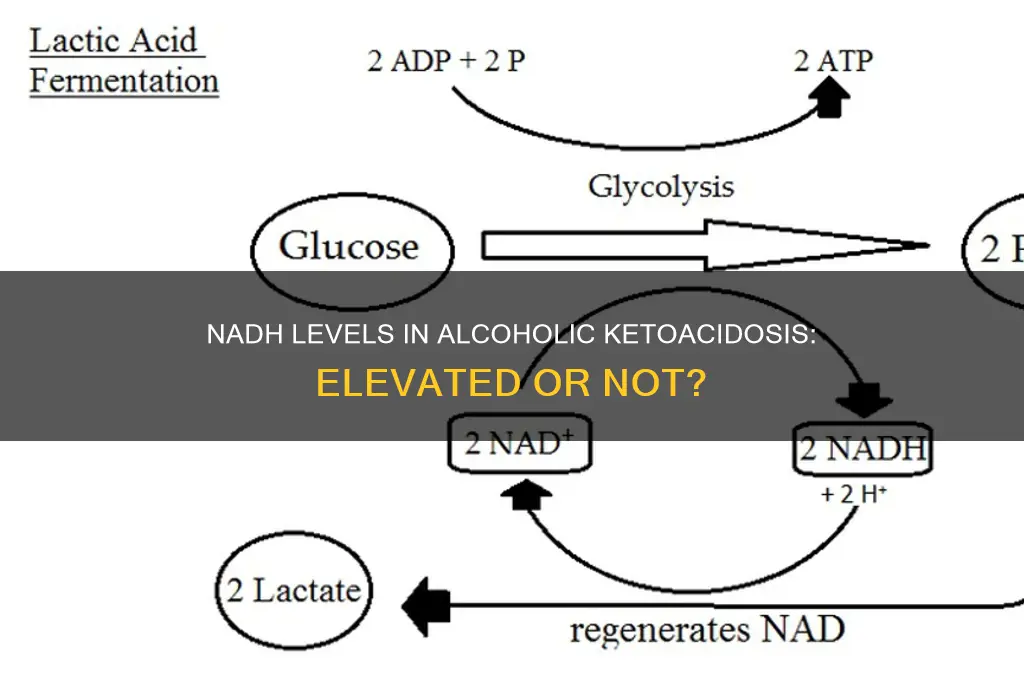
Nicotinamide adenine dinucleotide (NADH) is a critical coenzyme in cellular metabolism, acting as an electron carrier in redox reactions. Its role is particularly prominent in the electron transport chain, where it facilitates the transfer of electrons from nutrients to molecular oxygen, ultimately generating ATP. In the context of alcoholic ketoacidosis (AKA), understanding NADH’s function is essential, as ethanol metabolism disrupts its balance, leading to elevated levels. This elevation is not merely a byproduct but a key factor in the metabolic derangements observed in AKA.
Consider the metabolic pathway of ethanol: it is first converted to acetaldehyde by alcohol dehydrogenase, a reaction that reduces NAD+ to NADH. Subsequently, acetaldehyde is oxidized to acetic acid, further increasing NADH levels. This excessive NADH accumulation inhibits gluconeogenesis by impairing the conversion of pyruvate to oxaloacetate, a critical step requiring NAD+. As a result, the body shifts toward ketogenesis, producing ketone bodies that acidify the blood. For clinicians, recognizing this NADH-driven mechanism is crucial for diagnosing and managing AKA, especially in patients with a history of chronic alcohol consumption.
To mitigate NADH-related metabolic disruptions in AKA, targeted interventions can be employed. Administering thiamine (vitamin B1), a cofactor for pyruvate dehydrogenase, helps restore gluconeogenesis and reduce ketone production. Additionally, intravenous glucose replenishes glycogen stores and promotes the oxidation of NADH back to NAD+, rebalancing the redox state. For severe cases, dialysis may be considered to directly remove ketone bodies and correct acidosis. These strategies underscore the importance of addressing NADH elevation as a central component of AKA treatment.
A comparative analysis of NADH’s role in AKA versus diabetic ketoacidosis (DKA) reveals distinct differences. In DKA, NADH levels are typically not elevated; instead, the condition arises from insulin deficiency and counter-regulatory hormone excess. Conversely, AKA’s NADH elevation stems from ethanol metabolism’s direct interference with redox balance. This distinction highlights the need for tailored therapeutic approaches: while DKA requires insulin and fluid replacement, AKA demands attention to NADH modulation and nutrient repletion. Understanding these nuances ensures precise management of each condition.
Practically, monitoring NADH-related markers, such as the lactate-to-pyruvate ratio or venous blood gas analysis, can aid in early detection of AKA. Patients presenting with a history of alcohol use, nausea, and abdominal pain should prompt clinicians to investigate metabolic acidosis and ketonemia. Educating at-risk individuals about the dangers of chronic alcohol consumption and its metabolic consequences is equally vital. By focusing on NADH’s role, healthcare providers can adopt a proactive stance in preventing and treating alcoholic ketoacidosis effectively.
Alcohol Dependency Relapse: How Quickly Does It Return Post-Detox?
You may want to see also
Explore related products






![]()
Alcohol's Impact on NADH Levels
Alcohol consumption, particularly in excess, significantly disrupts the body's metabolic balance, leading to alterations in NADH (nicotinamide adenine dinucleotide) levels. NADH is a critical coenzyme in cellular respiration, facilitating the transfer of electrons and playing a pivotal role in energy production. When alcohol is metabolized, it competes with other substrates for the enzymes involved in NADH production, primarily in the liver. This competition results in an accumulation of NADH, tipping the NAD+/NADH ratio in favor of NADH. Such an imbalance is a hallmark of alcoholic ketoacidosis (AKA), a condition characterized by the buildup of ketone bodies due to impaired glucose utilization and increased fatty acid oxidation.
The mechanism behind NADH elevation in AKA begins with alcohol dehydrogenase (ADH) converting ethanol to acetaldehyde, a process that consumes NAD+ and produces NADH. Simultaneously, acetaldehyde is further metabolized by aldehyde dehydrogenase (ALDH), which also generates NADH. This dual pathway leads to a rapid depletion of NAD+ and a corresponding surge in NADH levels. For instance, chronic alcohol consumption can increase the NADH/NAD+ ratio by up to 10-fold in hepatic cells, according to studies. This imbalance impairs the Krebs cycle and oxidative phosphorylation, forcing the body to rely on ketogenesis for energy, ultimately leading to ketoacidosis.
From a practical standpoint, understanding the impact of alcohol on NADH levels is crucial for managing and preventing AKA. Clinicians often monitor this imbalance as a diagnostic marker, as elevated NADH levels correlate with the severity of metabolic acidosis in alcoholics. For individuals at risk, reducing alcohol intake is the primary intervention. Additionally, supplementation with NAD+ precursors, such as nicotinamide riboside, has shown promise in restoring the NAD+/NADH balance in preclinical studies. However, such interventions should be approached cautiously, as excessive supplementation can lead to adverse effects, including flushing and gastrointestinal distress.
Comparatively, the NADH elevation in AKA contrasts with conditions like diabetic ketoacidosis, where NADH levels are typically normal or slightly decreased. This distinction highlights the unique metabolic consequences of alcohol abuse. While both conditions result in ketoacidosis, the underlying mechanisms differ, emphasizing the need for tailored treatment strategies. For example, in AKA, thiamine supplementation is often prioritized due to its role in mitigating alcohol-induced metabolic dysfunction, whereas insulin therapy is central to managing diabetic ketoacidosis.
In conclusion, alcohol’s impact on NADH levels is a critical factor in the pathogenesis of alcoholic ketoacidosis. By disrupting the NAD+/NADH ratio, alcohol metabolism triggers a cascade of metabolic derangements, culminating in ketosis and acidosis. Recognizing this relationship not only aids in diagnosis but also informs targeted interventions, such as alcohol cessation and metabolic support. For those affected, early recognition and management of this imbalance can prevent life-threatening complications, underscoring the importance of addressing alcohol’s metabolic consequences in clinical practice.
Alcohol's Impact: Short-Term Memory and Low Doses
You may want to see also
Explore related products

$13.95





![]()
Ketoacidosis and NADH Elevation
NADH, or nicotinamide adenine dinucleotide (reduced), plays a critical role in cellular energy metabolism, acting as a key electron carrier in the electron transport chain. In the context of alcoholic ketoacidosis (AKA), a metabolic derangement often seen in chronic alcohol users, NADH levels become a focal point of interest due to the unique metabolic shifts that occur. Unlike diabetic ketoacidosis, where insulin deficiency is the primary driver, AKA is characterized by a depletion of NAD+ due to increased activity of alcohol dehydrogenase, the enzyme responsible for breaking down ethanol. This depletion of NAD+ subsequently leads to an elevation in NADH levels, as the oxidized form (NAD+) is consumed in the process.
To understand the implications of elevated NADH in AKA, consider the following metabolic pathway: ethanol is metabolized to acetaldehyde by alcohol dehydrogenase, a reaction that requires NAD+. Acetaldehyde is then further oxidized to acetic acid by aldehyde dehydrogenase, regenerating NAD+. However, in chronic alcohol consumption, this pathway becomes overwhelmed, leading to a significant reduction in NAD+ availability. The resulting increase in NADH disrupts the redox balance, impairing the cell’s ability to generate ATP efficiently. This metabolic imbalance contributes to the symptoms of AKA, including nausea, vomiting, abdominal pain, and, in severe cases, metabolic acidosis.
Clinically, addressing NADH elevation in AKA involves more than just fluid and electrolyte replacement. One practical intervention is the administration of thiamine (vitamin B1), a cofactor essential for the proper functioning of pyruvate dehydrogenase, an enzyme that helps convert pyruvate to acetyl-CoA. Thiamine deficiency, common in chronic alcohol users, exacerbates NADH accumulation by impairing this critical step in glucose metabolism. A typical thiamine replacement regimen includes an initial dose of 100–200 mg intravenously, followed by 50–100 mg daily until the patient’s nutritional status stabilizes. This approach not only helps restore metabolic balance but also mitigates the risk of complications such as Wernicke’s encephalopathy.
Comparatively, while both diabetic ketoacidosis (DKA) and AKA involve ketoacidosis, their underlying mechanisms and treatment strategies differ significantly. In DKA, insulin deficiency leads to increased lipolysis and ketogenesis, whereas in AKA, alcohol-induced metabolic shifts and NAD+ depletion are the primary culprits. This distinction highlights the importance of tailored treatment approaches. For instance, while insulin therapy is central to managing DKA, it is not required in AKA, where the focus is on replenishing NAD+ and correcting nutritional deficiencies. Recognizing these differences is crucial for clinicians to provide effective, condition-specific care.
In conclusion, elevated NADH in alcoholic ketoacidosis is a direct consequence of NAD+ depletion driven by chronic alcohol metabolism. This imbalance disrupts cellular energy production and contributes to the clinical manifestations of AKA. Practical interventions, such as thiamine supplementation and addressing nutritional deficiencies, are essential components of management. By understanding the unique metabolic dynamics of AKA, healthcare providers can implement targeted strategies to restore redox balance and improve patient outcomes. This nuanced approach underscores the importance of considering the specific biochemical pathways involved in metabolic disorders.
Liquid Benadryl and Alcohol: Uncovering the Truth About Ingredients
You may want to see also
Explore related products






![]()
NADH vs. NAD+ Ratio Changes
The NADH/NAD+ ratio is a critical metabolic marker, reflecting the balance between reductive and oxidative processes in cells. In alcoholic ketoacidosis (AKA), this ratio becomes dysregulated due to the unique metabolic demands and disruptions caused by chronic alcohol consumption and starvation. NADH levels are indeed elevated in AKA, primarily because ethanol metabolism depletes NAD+ through the activity of alcohol dehydrogenase, shifting the equilibrium toward NADH accumulation. This imbalance exacerbates metabolic acidosis and impairs cellular energy production, as NAD+ is essential for glycolysis and the citric acid cycle.
Analyzing the mechanism further, the conversion of ethanol to acetaldehyde by alcohol dehydrogenase consumes NAD+, leaving excess NADH. Simultaneously, fasting or starvation in AKA patients reduces glucose availability, forcing the liver to rely on fatty acid oxidation, which also generates NADH. This dual increase in NADH production, coupled with reduced NAD+ regeneration, creates a metabolic bottleneck. Clinically, this manifests as lactic acidosis and ketosis, as NADH inhibits key enzymes like lactate dehydrogenase and pyruvate dehydrogenase, diverting pyruvate away from oxidative metabolism and toward lactate production.
To address this imbalance, therapeutic strategies focus on restoring NAD+ levels. Intravenous thiamine (100–300 mg) is administered to cofactor-dependent enzymes, such as pyruvate dehydrogenase, which helps redirect metabolism toward oxidative pathways. Additionally, glucose infusion (1–2 mg/kg/min) replenishes glycogen stores and stimulates NAD+ regeneration via the pentose phosphate pathway. For severe cases, dichloroacetate (15–30 mg/kg/day) can be used to activate pyruvate dehydrogenase, bypassing NADH inhibition and reducing lactate levels. These interventions aim to normalize the NADH/NAD+ ratio, alleviating acidosis and metabolic dysfunction.
Comparatively, the NADH/NAD+ imbalance in AKA contrasts with conditions like diabetic ketoacidosis, where NAD+ depletion is less pronounced. In AKA, the combination of ethanol metabolism and starvation creates a unique metabolic milieu, making NAD+ restoration a cornerstone of treatment. Monitoring lactate and ketone levels can provide real-time feedback on the effectiveness of interventions, with a goal of reducing lactate below 2 mmol/L and resolving anion gap acidosis. Understanding this ratio’s dynamics not only clarifies the pathophysiology of AKA but also guides targeted therapy to restore metabolic homeostasis.
Mastering Alcohol Unit Counting: A Simple Guide to Track Your Intake
You may want to see also
Explore related products






![]()
Clinical Significance in Diagnosis
Elevated NADH levels in alcoholic ketoacidosis (AKA) serve as a critical diagnostic marker, offering clinicians a metabolic fingerprint of the disorder. This elevation stems from the excessive breakdown of ethanol, which prioritizes NADH production over NAD+ in the liver. The resultant NADH/NAD+ imbalance disrupts critical metabolic pathways, including gluconeogenesis and fatty acid oxidation, exacerbating ketone production and acidosis. Recognizing this biochemical shift allows clinicians to differentiate AKA from other causes of metabolic acidosis, such as diabetic ketoacidosis (DKA), where NADH levels typically remain stable.
Diagnostically, measuring NADH directly is impractical due to its labile nature and rapid turnover in vivo. Instead, clinicians infer NADH elevation through surrogate markers and clinical context. For instance, a patient presenting with a history of chronic alcohol use, metabolic acidosis with an elevated anion gap, and ketonemia in the absence of hyperglycemia should prompt suspicion of AKA. Laboratory findings such as hypokalemia, elevated liver enzymes, and a low serum bicarbonate level further support the diagnosis. The absence of significant hyperglycemia or hyperosmolarity helps distinguish AKA from DKA, where NADH levels do not play a central pathogenic role.
The clinical significance of NADH elevation extends beyond diagnosis, guiding therapeutic interventions. In AKA, the NADH/NAD+ imbalance impairs lactate metabolism, leading to lactic acidosis in some cases. Treatment strategies, such as the administration of intravenous thiamine (100 mg prior to glucose administration to prevent Wernicke’s encephalopathy) and hydration with isotonic saline, aim to restore metabolic homeostasis. Additionally, addressing the underlying ethanol toxicity by replenishing NAD+ through indirect means, such as promoting alcohol dehydrogenase activity, can help normalize NADH levels and mitigate acidosis.
A comparative analysis highlights the diagnostic utility of NADH elevation in AKA versus other conditions. While DKA relies on hyperglycemia-driven ketogenesis, AKA’s pathogenesis is tied to ethanol metabolism and NADH accumulation. This distinction underscores the importance of obtaining a thorough alcohol history and considering NADH-related metabolic derangements in patients with unexplained acidosis. For example, a 45-year-old male with a history of alcoholism presenting with tachypnea, abdominal pain, and a serum pH of 7.2 warrants immediate evaluation for AKA, with NADH-mediated metabolic disruption as a key diagnostic consideration.
In practice, clinicians should integrate knowledge of NADH elevation into a structured diagnostic approach. Begin with a focused history to identify alcohol consumption patterns, followed by laboratory tests including serum glucose, beta-hydroxybutyrate, and venous blood gas analysis. If AKA is suspected, prioritize thiamine supplementation before glucose administration to prevent precipitating Wernicke’s encephalopathy. This stepwise strategy, informed by the clinical significance of NADH elevation, ensures accurate diagnosis and timely intervention, improving outcomes for patients with this life-threatening condition.
Is the Legal Drinking Age Shifting? Exploring Global Trends and Debates
You may want to see also
Frequently asked questions
Yes, NADH levels are typically elevated in alcoholic ketoacidosis due to increased ethanol metabolism, which depletes NAD+ and shifts the balance toward NADH accumulation.
NADH elevation occurs because the breakdown of ethanol by alcohol dehydrogenase consumes NAD+ and produces NADH, leading to an imbalance in the NAD+/NADH ratio.
Elevated NADH impairs gluconeogenesis and promotes ketogenesis, contributing to the metabolic acidosis and ketosis seen in alcoholic ketoacidosis.
While NADH elevation is a metabolic consequence of alcoholic ketoacidosis, it is not typically measured clinically for diagnosis. Instead, symptoms, blood chemistry, and patient history are used.































