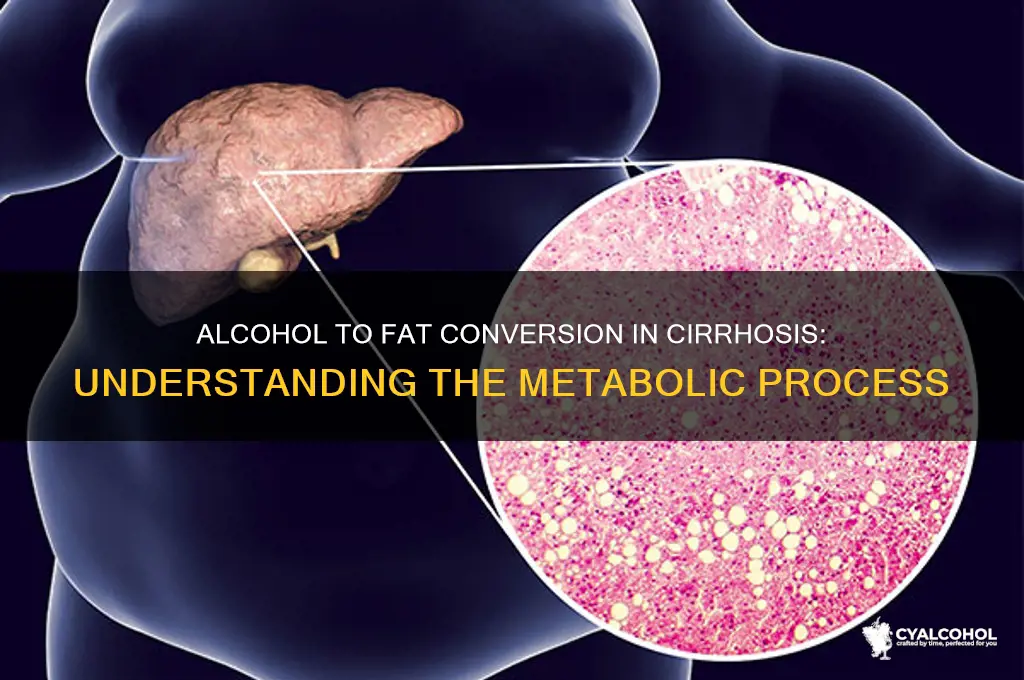
Alcohol-related cirrhosis is a severe liver condition characterized by extensive scarring and impaired liver function, often resulting from chronic alcohol consumption. In this disease, the liver’s ability to metabolize alcohol and fats is significantly compromised, leading to an abnormal accumulation of fat within liver cells. When alcohol is consumed, it is primarily metabolized in the liver by enzymes such as alcohol dehydrogenase and cytochrome P450 2E1, producing acetaldehyde and then acetate. However, in cirrhosis, this process is disrupted, and excess alcohol is shunted toward pathways that promote fat synthesis, such as increased production of NADH and acetyl-CoA, which are key intermediates in lipogenesis. Additionally, the damaged liver struggles to oxidize fatty acids, further exacerbating fat accumulation. This combination of enhanced fat production and reduced fat breakdown contributes to the development of fatty liver, a hallmark of alcoholic liver disease and a precursor to cirrhosis. Understanding this metabolic dysregulation is crucial for developing targeted therapies to mitigate the progression of alcohol-induced liver damage.
Explore related products





What You'll Learn
- Role of Acetaldehyde: Toxic byproduct damages liver cells, disrupts metabolism, promoting fat accumulation in cirrhotic livers
- Impaired Beta-Oxidation: Cirrhosis reduces fatty acid breakdown, leading to triglyceride buildup in liver cells
- Increased Lipogenesis: Alcohol induces enzymes like ACC, boosting fat synthesis in damaged liver tissue
- Insulin Resistance: Alcohol disrupts insulin signaling, enhancing fat storage and worsening cirrhosis-related steatosis
- Mitochondrial Dysfunction: Alcohol damages mitochondria, impairing energy production and increasing fat deposition in cirrhosis
![]()
Role of Acetaldehyde: Toxic byproduct damages liver cells, disrupts metabolism, promoting fat accumulation in cirrhotic livers
The process of alcohol metabolism plays a crucial role in the development of cirrhosis, a severe liver condition characterized by fat accumulation and scarring. When alcohol is consumed, the liver breaks it down into acetaldehyde, a highly toxic byproduct. This compound is short-lived but extremely reactive, and its presence initiates a cascade of detrimental effects on liver health. Acetaldehyde is primarily responsible for the liver damage associated with alcohol consumption, and its role in promoting fat accumulation in cirrhotic livers is a complex and multifaceted process.
In the liver, acetaldehyde exerts its toxic effects by damaging liver cells, known as hepatocytes. These cells are essential for various metabolic processes, including the breakdown and synthesis of fats. When acetaldehyde accumulates, it interferes with the normal functioning of hepatocytes, leading to cellular stress and damage. This damage triggers an inflammatory response, further exacerbating the liver's condition. The toxic byproduct also impairs the liver's ability to regulate fat metabolism, causing an imbalance in lipid handling. Normally, the liver processes and exports fats efficiently, but acetaldehyde disrupts this delicate system.
One of the key mechanisms by which acetaldehyde promotes fat accumulation is through the inhibition of lipid oxidation. Lipid oxidation is a vital process that breaks down fats into usable energy. However, acetaldehyde interferes with the enzymes responsible for this process, particularly carnitine palmitoyltransferase (CPT), which is essential for transporting fatty acids into the mitochondria for oxidation. As a result, fatty acids accumulate within the liver cells, leading to a condition known as steatosis, or fatty liver. This buildup of fat further compromises liver function and contributes to the progression of cirrhosis.
Additionally, acetaldehyde disrupts the normal metabolism of carbohydrates and lipids, favoring the synthesis of fatty acids over their breakdown. It stimulates the production of certain enzymes involved in lipogenesis, the process of converting excess carbohydrates into fats. This increased lipogenesis, coupled with impaired lipid oxidation, creates an environment conducive to fat accumulation. The liver becomes overwhelmed with fatty deposits, leading to the characteristic steatosis seen in alcoholic liver disease and cirrhosis.
The toxic effects of acetaldehyde also contribute to the development of insulin resistance, a condition where cells fail to respond properly to the hormone insulin. Insulin resistance is closely linked to fat metabolism disorders. In the context of cirrhosis, insulin resistance exacerbates fat accumulation by promoting increased lipid synthesis and decreased lipid breakdown. This metabolic dysfunction further aggravates the liver's ability to manage fat storage and utilization, creating a vicious cycle that accelerates the progression of liver damage. Understanding these mechanisms highlights the critical role of acetaldehyde in the pathogenesis of alcohol-induced liver disease and cirrhosis.
Alcohol and Pregnancy: Yolk Sac Protection
You may want to see also
Explore related products






![]()
Impaired Beta-Oxidation: Cirrhosis reduces fatty acid breakdown, leading to triglyceride buildup in liver cells
In the context of cirrhosis, impaired beta-oxidation plays a pivotal role in the accumulation of fat within liver cells, a process intricately linked to alcohol metabolism. Beta-oxidation is the primary pathway for breaking down fatty acids into acetyl-CoA, which can then enter the citric acid cycle to produce energy. However, chronic alcohol consumption disrupts this process, particularly in cirrhotic livers. Alcohol metabolism generates toxic byproducts like acetaldehyde and free radicals, which damage mitochondrial function—the site of beta-oxidation. This mitochondrial dysfunction impairs the liver’s ability to efficiently metabolize fatty acids, leading to their incomplete breakdown. As a result, fatty acids are not fully oxidized, and their intermediates accumulate, contributing to lipid buildup in hepatocytes.
Cirrhosis exacerbates this issue by further compromising liver function and mitochondrial integrity. The fibrotic and necrotic changes in cirrhotic liver tissue reduce the availability of functional hepatocytes capable of performing beta-oxidation. Additionally, alcohol-induced oxidative stress depletes essential cofactors such as carnitine, which is critical for transporting fatty acids into the mitochondria for oxidation. Without adequate carnitine, fatty acids cannot enter the beta-oxidation pathway, leading to their accumulation in the cytoplasm of liver cells. This buildup of fatty acids is then esterified into triglycerides, which are stored as lipid droplets within hepatocytes, contributing to fatty liver disease (steatosis).
The reduction in beta-oxidation efficiency is also linked to altered gene expression in cirrhotic livers. Chronic alcohol exposure downregulates the expression of genes encoding enzymes involved in fatty acid oxidation, such as carnitine palmitoyltransferase (CPT) and acyl-CoA oxidase. This genetic suppression further diminishes the liver’s capacity to break down fatty acids, perpetuating the cycle of lipid accumulation. Moreover, the inflammatory environment in cirrhosis, driven by alcohol-induced hepatocyte injury and immune cell activation, releases cytokines that inhibit beta-oxidation enzymes, compounding the problem.
Triglyceride buildup in liver cells due to impaired beta-oxidation is not merely a storage issue but has significant pathological consequences. Excessive lipid accumulation leads to hepatocyte ballooning, inflammation, and cell death, which are hallmark features of alcoholic liver disease. Over time, this progression from steatosis to steatohepatitis can contribute to fibrosis and cirrhosis, creating a vicious cycle of liver damage. Addressing impaired beta-oxidation through dietary interventions, such as carnitine supplementation or reducing alcohol intake, may help mitigate lipid accumulation and slow disease progression in cirrhotic patients.
In summary, impaired beta-oxidation in cirrhosis is a critical mechanism driving triglyceride buildup in liver cells. Chronic alcohol consumption damages mitochondrial function, depletes essential cofactors, and alters gene expression, all of which reduce the liver’s ability to break down fatty acids. This metabolic dysfunction leads to the accumulation of lipids within hepatocytes, exacerbating liver damage and disease progression. Understanding this pathway highlights the importance of targeting beta-oxidation impairment in the management of alcoholic liver disease.
Alcohol and Diarrhea: What's the Connection?
You may want to see also
Explore related products





![]()
Increased Lipogenesis: Alcohol induces enzymes like ACC, boosting fat synthesis in damaged liver tissue
Alcohol-induced liver damage, particularly in the context of cirrhosis, is closely linked to increased lipogenesis, a process where excess fat accumulates in liver cells. One of the key mechanisms driving this phenomenon is the induction of enzymes like acetyl-CoA carboxylase (ACC), which plays a pivotal role in fatty acid synthesis. When alcohol is metabolized in the liver, it disrupts normal cellular functions and alters metabolic pathways, leading to an upregulation of ACC activity. This enzyme catalyzes the conversion of acetyl-CoA to malonyl-CoA, a critical step in the synthesis of fatty acids. As a result, the liver begins to produce more fatty acids than it can effectively utilize or export, contributing to lipid accumulation.
The activation of ACC by alcohol is part of a broader metabolic shift in the liver. Chronic alcohol consumption increases the availability of acetyl-CoA, a byproduct of alcohol metabolism, which serves as a substrate for ACC. Additionally, alcohol impairs the liver's ability to oxidize fatty acids through mechanisms such as the inhibition of carnitine palmitoyltransferase 1 (CPT1), further exacerbating fat buildup. This imbalance between fatty acid synthesis and oxidation creates a favorable environment for increased lipogenesis, particularly in hepatocytes already damaged by alcohol. The damaged liver tissue becomes a site of excessive fat deposition, leading to conditions like alcoholic fatty liver disease (AFLD), which can progress to cirrhosis if left unchecked.
Another factor contributing to alcohol-induced lipogenesis is the activation of sterol regulatory element-binding protein 1 (SREBP-1), a transcription factor that upregulates genes involved in fatty acid and cholesterol synthesis, including ACC. Alcohol consumption promotes the maturation and nuclear translocation of SREBP-1, thereby enhancing the expression of lipogenic enzymes. This transcriptional activation further amplifies the liver's capacity for fat production, even in the presence of existing lipid overload. The interplay between alcohol metabolism, enzyme induction, and transcriptional regulation creates a vicious cycle that perpetuates fat accumulation and liver damage.
Moreover, the inflammatory response triggered by alcohol exacerbates lipogenesis in cirrhotic livers. Alcohol-induced inflammation increases the production of cytokines like tumor necrosis factor-alpha (TNF-α) and interleukin-6 (IL-6), which can stimulate ACC activity and promote fatty acid synthesis. This inflammatory milieu not only directly enhances lipogenesis but also impairs insulin signaling, leading to insulin resistance. Insulin resistance further upregulates ACC activity, as insulin normally suppresses this enzyme in healthy individuals. Thus, the combination of inflammation, insulin resistance, and enzyme induction creates a pro-lipogenic state that accelerates fat accumulation in damaged liver tissue.
In summary, increased lipogenesis in alcohol-induced cirrhosis is driven by the induction of enzymes like ACC, which are activated by alcohol metabolism, transcriptional regulation, and inflammatory pathways. The excessive production of fatty acids, coupled with impaired oxidation, results in lipid accumulation in hepatocytes, contributing to the progression of liver disease. Understanding these mechanisms highlights the importance of addressing alcohol consumption and metabolic dysregulation in the management and prevention of cirrhosis.
Home Alcohol Distilling in Iowa: What's the Law?
You may want to see also
Explore related products





![]()
Insulin Resistance: Alcohol disrupts insulin signaling, enhancing fat storage and worsening cirrhosis-related steatosis
Insulin resistance plays a pivotal role in the mechanism by which alcohol contributes to fat accumulation and worsens cirrhosis-related steatosis. Alcohol consumption disrupts insulin signaling pathways, impairing the hormone’s ability to regulate glucose and lipid metabolism effectively. Normally, insulin promotes glucose uptake by cells and suppresses the release of glucose from the liver. However, chronic alcohol intake interferes with insulin receptor function and downstream signaling, leading to reduced insulin sensitivity. This disruption causes cells to become less responsive to insulin’s effects, a hallmark of insulin resistance. As a result, the body compensates by producing more insulin, which further exacerbates metabolic imbalances.
One of the direct consequences of insulin resistance induced by alcohol is the enhancement of fat storage. Insulin normally inhibits lipolysis (the breakdown of fats) in adipose tissue and stimulates lipogenesis (fat synthesis) in the liver. When insulin signaling is disrupted, this balance is lost. The liver, in particular, becomes a site of excessive fat accumulation due to increased lipogenesis and reduced fatty acid oxidation. Alcohol metabolism generates acetyl-CoA, a precursor for fatty acid synthesis, which further fuels this process. Additionally, insulin resistance promotes the release of free fatty acids from adipose tissue, which are then transported to the liver, contributing to hepatic steatosis.
The interplay between alcohol-induced insulin resistance and fat accumulation creates a vicious cycle that worsens cirrhosis-related steatosis. Hepatic steatosis, characterized by the buildup of fat in liver cells, is an early stage of alcoholic liver disease. As insulin resistance persists, the liver’s ability to manage lipid metabolism deteriorates, leading to progressive fat deposition. This accumulation of lipids in hepatocytes triggers inflammation and oxidative stress, which are key drivers of liver damage. Over time, this can progress to fibrosis, cirrhosis, and eventually liver failure if alcohol consumption continues unchecked.
Furthermore, insulin resistance exacerbates cirrhosis by impairing the liver’s regenerative capacity and promoting hepatocyte apoptosis. Elevated insulin levels and dysregulated lipid metabolism contribute to the production of pro-inflammatory cytokines, which further damage liver tissue. The combination of fat accumulation, inflammation, and cellular stress creates an environment conducive to the progression of liver disease. Addressing insulin resistance through lifestyle modifications, such as reducing alcohol intake and improving dietary habits, is crucial for mitigating these effects and slowing the advancement of cirrhosis.
In summary, alcohol disrupts insulin signaling, leading to insulin resistance that enhances fat storage and worsens cirrhosis-related steatosis. This process involves impaired glucose and lipid metabolism, increased hepatic lipogenesis, and reduced fatty acid oxidation. The resulting fat accumulation in the liver, coupled with inflammation and oxidative stress, accelerates the progression of liver disease. Understanding this mechanism underscores the importance of managing insulin resistance and alcohol consumption in the prevention and treatment of alcoholic liver disease.
Drugged Driving: The Other Impaired Driving Danger
You may want to see also
Explore related products






![]()
Mitochondrial Dysfunction: Alcohol damages mitochondria, impairing energy production and increasing fat deposition in cirrhosis
Alcohol-induced liver damage, particularly in the context of cirrhosis, is closely linked to mitochondrial dysfunction, a critical process that exacerbates fat deposition in the liver. Mitochondria, often referred to as the "powerhouses" of the cell, play a pivotal role in energy production through oxidative phosphorylation. However, chronic alcohol consumption directly damages mitochondrial structure and function. Alcohol metabolites, such as acetaldehyde and reactive oxygen species (ROS), disrupt mitochondrial membranes, impairing their ability to generate adenosine triphosphate (ATP). This energy deficit forces hepatocytes to shift toward alternative energy sources, promoting lipid accumulation.
One of the key mechanisms by which mitochondrial dysfunction contributes to fat deposition is the inhibition of β-oxidation, the primary pathway for fatty acid breakdown. Alcohol-damaged mitochondria exhibit reduced expression and activity of enzymes critical for β-oxidation, such as carnitine palmitoyltransferase 1 (CPT1). As a result, fatty acids cannot be effectively metabolized, leading to their accumulation within hepatocytes. This intracellular lipid buildup is a hallmark of alcoholic fatty liver disease (AFLD), the initial stage of alcohol-induced liver injury that can progress to cirrhosis if left unchecked.
Additionally, mitochondrial dysfunction disrupts the balance between lipid synthesis and degradation. Alcohol increases the activity of sterol regulatory element-binding protein 1c (SREBP-1c), a transcription factor that upregulates genes involved in fatty acid and triglyceride synthesis. Simultaneously, impaired mitochondria fail to provide sufficient ATP for proper lipid export, further contributing to fat accumulation. This dual effect—enhanced lipid production and reduced lipid clearance—creates a vicious cycle that accelerates hepatic steatosis, a precursor to cirrhosis.
Another critical aspect of mitochondrial dysfunction is the overproduction of ROS, which not only damages mitochondrial DNA and proteins but also activates stress-responsive pathways that promote lipogenesis. ROS-induced oxidative stress triggers the activation of AMP-activated protein kinase (AMPK), a cellular energy sensor that, when inhibited, reduces fatty acid oxidation and increases lipid storage. Furthermore, ROS activates tumor necrosis factor-alpha (TNF-α), a pro-inflammatory cytokine that exacerbates liver injury and fat deposition by impairing insulin signaling and promoting lipid synthesis.
In summary, mitochondrial dysfunction is a central mechanism by which alcohol consumption leads to fat deposition in cirrhosis. By impairing energy production, inhibiting fatty acid oxidation, disrupting lipid homeostasis, and inducing oxidative stress, alcohol-damaged mitochondria create an environment conducive to hepatic steatosis. Understanding these processes highlights the importance of mitochondrial health in preventing and treating alcohol-induced liver disease, emphasizing the need for therapeutic strategies targeting mitochondrial function to mitigate fat accumulation and disease progression.
Altitude and Alcohol: Denver's Drinking Culture
You may want to see also
Frequently asked questions
Alcohol disrupts the liver's normal metabolic processes, leading to increased fat production and reduced fat breakdown. This results in excessive fat accumulation, a condition known as alcoholic fatty liver, which can progress to cirrhosis over time.
Acetaldehyde, a toxic byproduct of alcohol metabolism, impairs the liver's ability to process fats. It increases the production of fatty acids and triglycerides while inhibiting their oxidation, contributing to fat buildup and liver damage in cirrhosis.
Alcohol consumption promotes insulin resistance, which disrupts glucose and fat metabolism. This leads to increased fat storage in the liver and reduced fat mobilization, exacerbating fatty liver disease and progressing to cirrhosis.
Yes, early-stage alcohol-induced fat accumulation (alcoholic fatty liver) can often be reversed with abstinence from alcohol and lifestyle changes. However, if left untreated, it can progress to cirrhosis, which is irreversible.
Chronic alcohol use causes sustained fat buildup in the liver, leading to inflammation and scarring (fibrosis). Over time, repeated liver damage results in cirrhosis, a severe condition characterized by irreversible liver scarring and dysfunction.































